教程
Gaussian专题
Gaussian09首次使用配置教程
在Linux上运行Gaussian
Gaussian输入文件模板
Gaussian中chk文件的相关问题
Gaussian结构优化相关问题
提交Gaussian任务后立即报错的解决方法
guassian+multiwfn计算FuKui福井函数
Gaussian+Multiwfn算极化率相关值
Pandas中DataFrame的创建与取值
Linux常用命令
生成forcite脚本
命令行提交castep任务的教程
ms中保证同一系列任务参数一致的方法
在MaterialsStudio中使用多核并行Amorphous Cell模块的方法
MS 建模中常用命令的快捷键
Linux中vi文本编辑器的命令
Multiwfn+VMD轨道图绘制
TensorFlow CPU版本安装
Jupyter notebook使用
MS中导出高质量图片的教程
Oringin作图合集
oringin画反应能级图数据模板
Oringin自定义非均匀横坐标+添加参考线
forcite模块RDF&MSD教程
Materials Studio连接服务器
Clash教程
分数坐标转直角坐标excel
liuyaoze.com-文档系统
-
+
首页
guassian+multiwfn计算FuKui福井函数
#### **参考链接:** [使用Multiwfn超级方便地计算出概念密度泛函理论中定义的各种量](http://sobereva.com/484 "使用Multiwfn超级方便地计算出概念密度泛函理论中定义的各种量") [使用Multiwfn作电子密度差图](http://sobereva.com/113 "使用Multiwfn作电子密度差图") ==Fukui(福井)函数==是常用的预测反应位点的实空间函数,尤其适合软酸软碱之间的反应(静电势的分析方法更适合用于分析硬酸硬碱反应)。Fukui函数有三类,分别用于预测亲核、亲电、自由基反应位点。预测亲电反应的函数f- = ρ(N) - ρ(N-1),其中ρ(N)是分子正常状态下的密度函数,ρ(N-1)是分子少了一个电子,或者说分子的净电荷为+1状态下的密度。f-在哪个原子附近数值越大,说明亲电反应越容易发生在哪个原子上。注意计算ρ(N-1)时几何结构必须使用计算ρ(N)时的,这不仅是绘制密度差图的要求,这也是Fukui函数的定义本身所要求的。 ### 1.对结构进行优化 选择合适的基组 例: >d 注意,所有输入文件仅作为关键词参考,记得保持文件名,chk文件名,提交任务脚本中的文件名,fchk文件名的统一,末尾空两行,title上下空两行 ``` %chk=N.chk %nprocshared=24 # opt freq ub3lyp 6-31(d,p) Title Card Required 0 1 S -0.43000000 0.58200000 0.51600000 O 0.05000000 -1.13100000 0.50800000 O -0.30800000 1.19600000 2.18100000 N 0.74600000 1.48900000 -0.46200000 C 2.21900000 1.19600000 -0.29000000 C 3.37500000 1.98600000 -0.90000000 C 4.66000000 1.35700000 -0.36000000 O 4.28600000 0.20900000 0.55500000 N 2.79400000 0.10600000 0.59300000 H 0.40400000 2.25500000 -1.18800000 C -2.15600000 0.70500000 -0.00400000 C -2.86700000 -0.53400000 -0.57600000 C -4.38600000 -0.50500000 -0.81300000 C -5.18800000 0.76700000 -0.48300000 C -4.46600000 2.02900000 0.02100000 C -2.94700000 1.99800000 0.26400000 N -6.68600000 0.77400000 -0.65200000 C 6.08200000 1.92800000 -0.52300000 H 3.29300000 2.89400000 -1.58100000 H -2.27300000 -1.48100000 -0.79600000 H -4.91800000 -1.42600000 -1.22000000 H -5.05700000 2.98000000 0.22900000 H -2.41400000 2.92100000 0.66500000 H -7.20800000 -0.13700000 -1.01400000 H -7.26600000 1.68900000 -0.41400000 H 6.57800000 1.45800000 -1.43500000 H 6.69100000 1.68100000 0.40600000 H 6.03400000 3.05900000 -0.65400000 ``` 如果发现没有电荷但是自旋多重度不为1,可能是分子式没配平,会出现l301报错 ### 2.计算波函数文件 使用上一步优化好的结构,计算波函数文件 这是不外加电荷的正常情况 ``` %chk=N.chk %nprocshared=24 # b3lyp/6-31(d,p) out=wfn opted 0 1 S 0.13649800 1.09980500 0.11075100 O -0.08658700 1.91318900 -1.07899500 O -0.05754400 1.65197900 1.45581200 N -0.81257200 -0.34311300 -0.07751200 C -2.21555300 -0.28955900 -0.10677600 C -3.07866800 -1.33259700 0.35109100 C -4.32467600 -0.85895700 0.07437900 O -4.22990200 0.35905900 -0.49480500 N -2.87768600 0.72526400 -0.61768300 H -0.47101400 -1.04498700 0.57237600 C 1.74884400 0.34815600 0.04417700 C 2.26964900 -0.06458300 -1.18766000 C 3.53629900 -0.62456700 -1.24416300 C 4.30907200 -0.77401800 -0.07300600 C 3.77151900 -0.34224400 1.15578900 C 2.50194400 0.21687700 1.21402800 N 5.55275700 -1.37425500 -0.12561700 C -5.69603000 -1.40363200 0.27434100 H -2.80862500 -2.27320800 0.80633000 H 1.68454200 0.06495300 -2.09171300 H 3.94322900 -0.94776600 -2.19834900 H 4.36001800 -0.44415000 2.06343500 H 2.09228200 0.56671700 2.15535500 H 6.01381800 -1.39373700 -1.02321000 H 6.17882200 -1.19834900 0.64622800 H -6.21945100 -1.49948600 -0.68247000 H -6.28885900 -0.73836300 0.91030200 H -5.64835400 -2.38685800 0.74609100 ``` 就可以得到N.fchk文件(使用 formchk a.chk a.fchk命令生成,a为文件名) > N指的是体系的电子数,比如某个分子最稳定状态是中性的,则N就是指中性时的电子数,N+1就是指带一个负电荷的阴离子状态的电子数,N-1就是指带一个正电荷的阳离子态的电子数。 算完后把净电荷和自旋多重度从0 1改成1 2,N.fchk改成N-1.fchk,重算一遍得到N-1电子时的波函数文件,同理改成-1 2,计算多一个电子的N+1.fchk ### 3.计算福井函数 现在当前目录下已经有了N.fchk、N+1.fchk和N-1.fchk,可以开始算了。 随意拖一个fchk文件进Multiwfn,回车 > 三个态的波函数文件也不是必须叫做N.wfn、N+1.wfn和N-1.wfn并处在当前目录下。后面开始计算时,若Multiwfn发现当前目录下缺少这些文件的一个或多个,程序就会让用户直接输入相应电子态的.wfn或.wfx或.fch或.mwfn文件的路径。因此,比如你想用自行算出来的N.fch、N+1.fch、N-1.fch做后面的计算也完全可以。 22 //计算CDFT里定义的各种量 1 //产生波函数文件 [直接按回车] //这一步让你输入gjf文件里对应的计算单点任务的关键词。按回车代表用默认的B3LYP/6-31G* [直接按回车] //这一步让你输入计算N、N+1和N-1态用的电荷和自旋多重度。按回车代表用对三个态分别用默认的0 1、-1 2和1 2 2 // Calculate various quantitative indices,然后程序就会依次载入fchk文件,读取其中的能量信息和波函数信息,自动计算Hirshfeld电荷,最后给出各种指数。算完的数据被输出到了当前目录下的CDFT.txt中,此文件内容如下 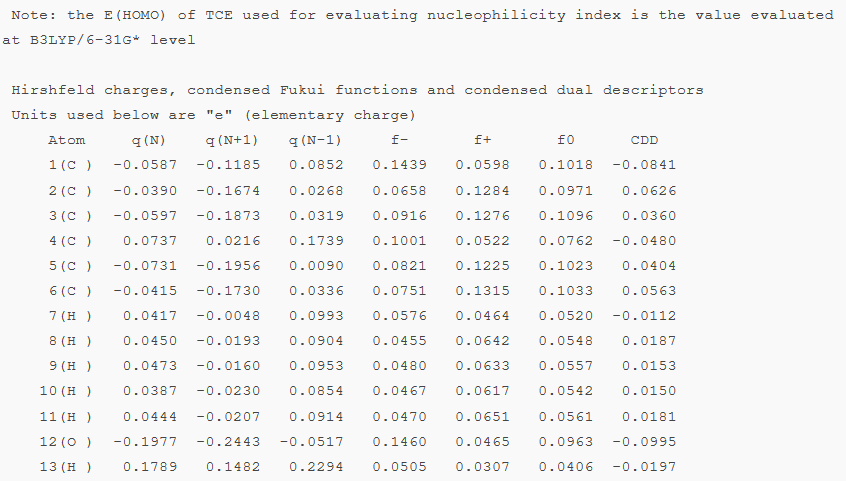 接下来计算福井函数和双描述符的等值面图,在当前模块里选择 3 //Calculate grid data of Fukui function and dual descriptor,选择一个合适的格点设定,对于当前这样小体系选择Medium quality grid就够了(大体系应当用High quality grid或其它选项) 然后程序会依次载入当前目录下的N.wfn、N+1.wfn和N-1.wfn并计算电子密度格点数据 之后会看到一个菜单,通过选择相应选项,可以把各种类型的福井函数以及双描述符直接显示成等值面图。比如此例我们依次选择选项1、2、3、4来把f+、f-、f0福井函数和双描述符都依次绘制出来,下图是把图像手动合并到一起的图,等值面数值都是用的0.007: 
huanganqi
2023年3月1日 22:14
转发
收藏文档
上一篇
下一篇
手机扫码
复制链接
手机扫一扫转发分享
复制链接
【温馨提示:本站文档可配置可见范围,如登录后可见、对特定群组可见等,看不到或进不去就是没权限】
暂时不开放注册
Markdown文件
Word文件
PDF文档
PDF文档(打印)
分享
链接
类型
密码
更新密码
有效期